The Internal Standard Method in Gas-Liquid Chromatography
David L. Zellmer
Department of Chemistry
California State University, Fresno
May 6, 1998
The need for an Internal Standard can be illustrated with a simple spreadsheet exercise.
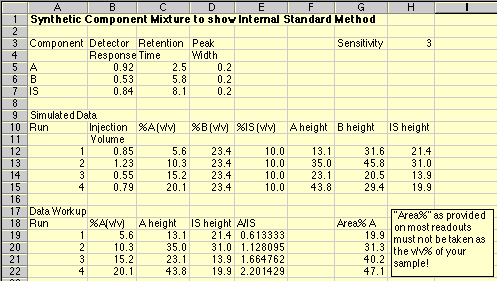
Note that components "A", "B" and "IS" each have their own response to the detector, as well as distinct retention times. This simulation uses constant peak widths to make the peak areas proportional to the peak height, simplifying the calculations needed.
A series of Standard Solutions has been prepared, each having a different amount of "A" in it. The volume% of "B" and of the Internal Standard "IS" is held constant in each sample. As each sample is run, the Hamilton syringe is unable to inject a constant 1.00 microliter volume; the actual amounts injected are given in the Injection Volume column. In real life you would not know what these values are. Note that run 3 had a particularly bad problem, with only about half of the intended sample being placed on the column.
The final peak height of "A" is the product of (Injection Volume)*(%Av/v)*(Detector Response A)*(Sensitivity). The Sensitivity is just a number to make the peak heights come out to a nice size. It is similar to the sensitivity or attenuation settings we use in the lab to get our peaks to come out the right size on our readouts.
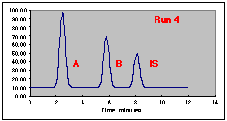
The following chromatograms are the result. Note that our "A" peak is not increasing with concentration as we would have hoped.
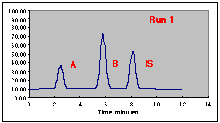
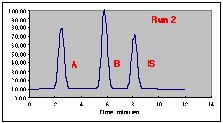
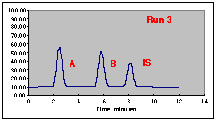
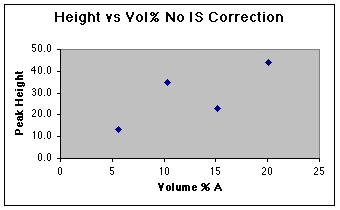
A working curve of Peak Height vs. Volume Percent confirms our worst fears.
But wait! All is not lost. By dividing the "A" peak height by the height of the Internal Standard (IS) we can correct for the varying injection volumes.
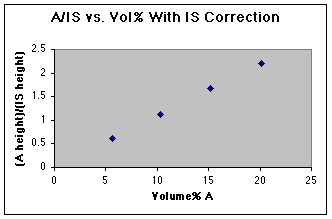
We now have a working curve we can use. Had an unknown been run as well, the unknown A/IS could be used to read the volume% A from this graph.
Determining an Unknown Concentration of "A"
Assume an unknown is prepared by taking 50.0 mL of unknown, 10.0 mL of IS, and diluting to 100.0 mL with solvent. The following chromatogram is produced.
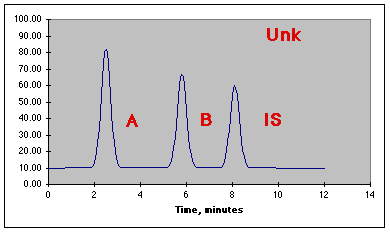
The data from the run is:
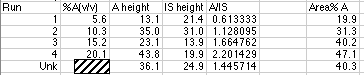
We have blocked out the volume% A used to prepare the simulated peaks for the unknown. Linear Least Squares analysis will be used to calculate this from our working curve.
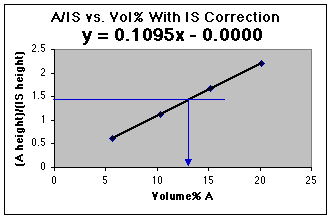
Using the LLS line, and our value of A/IS for the unknown, we find that 1.446 = 0.110 x - 0.000. Solving for x, we get (1.446 - 0.000)/0.1095 = 13.2 volume %, which is correct for your diluted sample. Don't forget that the original unknown has been diluted from 50.0 mL to 100.0 mL, so the original unknown was 13.2 volume % x 100 mL/50 mL = 26.4 volume %. For real world analyses you should use the more complex LLS analysis which includes calculation of the error of the concentration found on the x-axis.
Errors to Avoid
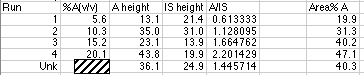
Chromatographic data stations and integrators will give you a report on your chromatograph that includes things like Retention Time, Peak Area, and Area%. The Area% of "A" is simply the area of peak A divided by the sum of the areas of all the other peaks x 100. A very naive person might think that the Area% is somehow magically equal to the Volume% they are trying to measure in the unknown material. "The machine said it was 38.3345 %," is only said by people who have seen too many episodes of Star Trek where Tricorders provide instant analyses of anything. Note that in our simulation, the Area % bears no resemblance to the Volume % of our standards.
Our simulated unknown is certainly not 40.3 volume%. We couldn't even use a plot of Area% A vs. concentration to get a valid working curve. Inspection of the data above shows that such a plot would give a value of around 15 volume% for our unknown, when its real value was 13.2 volume%. It happened to be somewhat close, but not correct.